Oligomerization of a peptide was attempted in a flow reactor that simulated a submarine hydrothermal system. When fluid containing glycine repeatedly circulated through the hot and cold regions in the reactor, oligopeptides were made from glycine. When divalent ions (such as copper ions) were added under acidic conditions, oligoglycine was elongated up to hexaglycine. This observation suggests that prebiotic monomers could have oligomerized in the vicinity of submarine hydrothermal vents on primitive Earth.
* To whom correspondence should be addressed.
E-mail: kmatsuno@vos.nagaokaut.ac.jp
The onset of polymerization must have been a major step in the chemical evolution that formed the precursors of life (1-4). The underlying chemical reaction requires an organization in which products can be repeatedly transformed into reactants, as seen with ribosomes and ribozymes in contemporary biological organisms. Systems or processes that could have assisted the transformation of products to reactants might include heating in dry and wet conditions, the diurnal cycle, tidal waves, and dry-wet cycles in lagoons (5). Submarine hydrothermal vents (6) have been recognized as a possible environment for prebiotic synthesis; in this environment, products that were synthesized in hot vents could reenter the vents after being quenched in the surrounding cold water.
The thermal synthesis of products in hot vents (7) and the subsequent rapid cooling in surrounding cold water are generative and selective when combined (8). Thus, hydrothermal vents in the sea could have been an environment where oligomers and polymers were synthesized and selected. For instance, when two amino acid molecules form a peptide bond in hot vents and then the product is ejected into the surrounding cold water, the peptide bond could survive in the cold environment if the dissociation process [including decarboxylation, deamination, or dehydration (9)] is retarded.
We constructed a flow reactor that simulated the pressure and temperature conditions of the hydrothermal circulation of water in order to examine the likelihood of synthesizing oligopeptides from monomeric amino acids (10). However, there were still some large differences, for instance, in pH, CO2, Na, and Cl contents. In our flow reactor (Fig. 1), a high-temperature high-pressure fluid was injected into a low-temperature chamber that was maintained at about the same high pressure as the fluid. The fluid circulated in a closed manner in the system with a fixed turnover rate. The fluid was heated and compressed in one part of the circuit; the rest of the chamber was cooled externally. Samples of the fluid were repeatedly taken from the low-temperature chamber for measurement at a given time interval, and the fluid in the low-temperature chamber was then returned into the high-temperature high-pressure fluid.
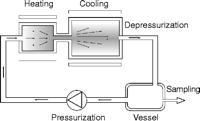
Fig. 1. A schematic drawing of a flow reactor
simulating a submarine hydrothermal system.
We prepared 100 mM glycine solution that was dissolved in pure water, and we maintained the total volume of the circulating fluid at 500 ml. The pressure of the high-pressure high-temperature chamber with its 15-ml volume was set at 24.0 MPa, which is only slightly higher than the pressure of the critical point of water (22.1 MPa). This pressure was chosen to maintain the water in the chamber as a liquid. The temperature of the high-temperature chamber was varied from 110° to 350°C in different runs. The results of interest were obtained for temperatures ranging roughly between 200° and 250°C. Temperature was increased gradually over 20 min. We started the measurements of the yields when the designated temperature was reached. The diameter of the nozzle from which a jet stream of high-temperature high-pressure fluid was injected into the low-temperature chamber was 100 µm, and the injection rate was 8 to 12 ml/min. The chamber had a fluid volume of 78.5 ml, and the temperature was maintained externally at 0°C. The flow rate of the jet stream was adjusted to maintain the pressure of the low-temperature chamber at 24.0 MPa. Accordingly, the turnover time of the whole circulating fluid was 1.0 to 1.3 hours. The downstream vessel was depressurized by inserting a stainless steel tube (0.5 m long and 100 µm in diameter) between the high-pressure low-temperature chamber and the depressurized vessel. For measurements, aliquots of 5 µl were taken out of the depressurized vessel at fixed time intervals.
The reacting chemicals cycled from the low-temperature chamber back to the high-temperature chamber in either 34 or 78 s, which is much shorter than the turnover time of the whole fluid. The cycle time was set by inserting different lengths of tube (800 µm in diameter) between the depressurized vessel and the pressurization pump. It was confirmed that reactants in the different vessels were stirred (11). The major factor determining the cycle time of reactants was the convection of the fluid through the 100-µm tube.
The temperature of the injecting jet stream from the nozzle that was connected to the high-pressure high-temperature chamber was first set at 225°C; the reaction fluid contained 100 mM glycine that was dissolved in pure water with no pH control or added salts. A high-performance liquid chromatography (HPLC) profile of the products revealed that, with time, at least three different oligomers formed: diketopiperazine and the dimer and trimer of glycine (Fig. 2, A and B). We also identified these species with LC mass spectroscopy. The initial growth of both the dimer and the trimer was exponential in time. The doubling time was 33 s for the cycle time of 34 s and was 80 s for the cycle time of 78 s (Fig. 2C). The coincidence between the doubling time and the cycle time of reactants traveling the closed path of the flow reactor suggests that both di- and triglycine formed sequentially, in the sense that the preceding products served as a base for adding monomers one by one as compounds repeatedly traveled the closed path of the reactor.
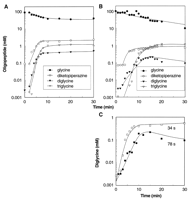
Fig. 2. Time courses of the reaction yields for the
reaction fluid containing only 100 mM glycine, with no pH control or added salts. The
reactants traveled the closed path of the flow reactor from the high-pressure
low-temperature chamber back to the high-pressure high-temperature chamber in cycle times
of (A) 34 and (B) 78 s. For reference, the amount of monomeric
glycine in the solution is also presented. (C) Time development of the yields of
diglycine for cycle times of 34 and 78 s. The yield was estimated by referring
the area of each corresponding HPLC peak to a standard reference of a given concentration.
The linearity between the area and the concentration was confirmed. The temperature of the
high-pressure high-temperature chamber was set at 225°C. All samples were analyzed with a
Hitachi (L-6300, L-4200, and D-2500) HPLC apparatus with a Shodex Asahipak column (ODP-50)
(5 µm by 4.6 mm by 150 mm). The mobile phase consisted of 50 mM KH2PO4
and 7.2 mM C6H13SO3Na, and its pH was maintained at
2.5 by adjusting the added amount of H3PO4. The flow rate of
the mobile phase was 0.5 ml/min; this was detected by measuring the absorbance at
195 nm. As standards, glycine and its oligomers up to hexaglycine were purchased from
Sigma-Aldrich.
When 10 mM CuCl2 was added to the 100 mM glycine solution and the pH was adjusted to 2.5 by HCl at room temperature, higher oligomers were obtained (Fig. 3) in an experiment in which the temperature of the high-pressure high-temperature chamber was set at 250°C at 24.0 MPa and the cycle time was maintained at 34 s. A HPLC profile identified at least four different oligomers: diketopiperazine, diglycine, tetraglycine, and hexaglycine (12). Copper ions were found to help synthesize tetraglycine, as suggested by its exponential initial growth. Even hexaglycine was synthesized after a sufficient amount of tetraglycine was formed.
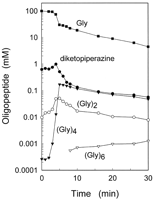
Fig. 3. Time courses of the reaction yields for the reaction fluid that
contained 100 mM glycine and 10 mM CuCl2 and was adjusted to a pH of
2.5 by HCl at room temperature. The temperature of the high-pressure high-temperature
chamber was set at 250°C at 24.0 MPa. The HPLC conditions were the same as in Fig. 2. For reference, the amount of monomeric glycine (Gly) in the
solution is also presented; (Gly)2 represents diglycine, (Gly)4
represents tetraglycine, and (Gly)6 represents hexaglycine.
The fact that di- and triglycine were synthesized with no detectable amount of tetraglycine in pure water suggests that tetraglycine molecules could be rapidly hydrolyzed into two molecules of diglycine. Two molecules of diglycine could then yield two more molecules of triglycine when they reentered the reaction region in the high-pressure high-temperature chamber. The initial increment of the yields of triglycine also suggests that monomeric glycine could aminolyse diketopiperazine to form triglycine (13).
The presence of copper ions seems to have prevented the hydrolysis of tetraglycine. Tetraglycine therefore reentered the reaction region and further reacted with a glycine, producing a diglycine, a triglycine, or a diketopiperazine molecule when the amount of tetraglycine becomes sufficient. The presence of even-numbered oligomers up to hexaglycine and the absence of detectable amounts of both tri- and pentaglycine suggest that the chain elongation proceeds mainly by aminolysis of diketopiperazine.
As monomers of biological significance, both amino acid and nucleotide molecules can potentially accommodate stepwise polymerization schemes into themselves (2, 3) [for instance, by repeating the cycle of hydrolysis and elongation (4)]. From an evolutionary perspective, a more pressing issue in this regard is how to implement such schemes. Stepwise synthesis of oligoglycine in our flow reactor seems to suggest that submarine hydrothermal vents in the Archean ocean could have readily facilitated the multiplicative oligomerization of these monomers, even in the absence of ribosomes or ribozymes.
REFERENCES AND NOTES
- M. Eigen, Naturwissenschaften 58, 465 (1971) [ISI][Medline] .
- K. Matsuno, BioSystems 15, 1 (1982) [ISI][Medline].
- S. A. Kauffman, J. Theor. Biol. 119, 1 (1986) [ISI][Medline] .
- L. E. Orgel, ibid. 123, 127 (1986) [ISI][Medline].
- T. Nakashima, J. R. Jungck, S. W. Fox, D. Lederer, B. C. Das, Int. J. Quantum Chem. QBS4, 65 (1977) ; C. Huber and G. Wächtershäuser, Science 281, 670 (1998) [Abstract/Full Text] ; M. Keller, E. Blöchl, G. Wächtershäuser, K. O. Stetter, Nature 368, 836 (1995) [ISI] ; A. Brack, Origins Life Evol. Biosphere 17, 367 (1987) [ISI][Medline] ; B. M. Rode and G. Schwendinger, ibid. 20, 401 (1990) [ISI]; G. Wächtershäuser, Prog. Biophys. Mol. Biol. 58, 85 (1992) [ISI][Medline] ; J. Bujdak, K. Faybikova, A. Eder, Y. Yongyai, B. M. Rode, Origins Life Evol. Biosphere 25, 431 (1995) [ISI][Medline] ; L. E. Orgel, ibid. 28, 227 (1998) [ISI][Medline]; J. P. Ferris, A. R. Hill Jr., R. Liu, L. E. Orgel, Nature 381, 59 (1996) [ISI][Medline] ; A. D. Keefe, G. L. Newton, S. L. Miller, ibid. 373, 683 (1995) [ISI][Medline]; M. P. Robertson and S. L. Miller, ibid. 375, 772 (1995).
- J. B. Corliss, et al., Science 203, 1073 (1979) [ISI] ; J. M. Edmond, K. L. Von Damm, R. E. McDuff, C. I. Measures, Nature 297, 187 (1982) [ISI] ; H. Yanagawa and K. Kojima, J. Biochem. 97, 1521 (1985) [ISI][Medline] ; M. J. Russell, A. J. Hall, A. G. Cairns-Smith, P. S. Braterman, Nature 336, 117 (1988) [ISI] ; E. L. Shock, Ciba Found. Symp. 202, 40 (1996) [ISI][Medline] .
- J. P. Ferris, Origins Life Evol. Biosphere 22, 109 (1992) [ISI][Medline] ; J. P. Amend and E. L. Shock, Science 281, 1659 (1998) [Abstract/Full Text] .
- K. Matsuno, Protobiology: Physical Basis of Biology (CRC Press, Boca Raton, FL, 1989). When a small hot body comes into contact with a large cold body, the temperature of the hot body decreases. In particular, the actual temperature drop is the fastest possible alternative. If the slower process was to take over, the heat flow toward the cold body would increase because of the greater temperature difference between the two bodies. The greater transference of heat energy toward the cold body with a smaller temperature drop of the hot body in time is, however, a self-contradiction. Actualization of the fastest temperature drop is selective in comparison to generative thermal synthesis at a high temperature.
- J. L. Bada, S. L. Miller, M. Zhao, Origins Life Evol. Biosphere 25, 111 (1995) [ISI][Medline] .
- K. Matsuno, Viva Orig. 25, 191 (1997) . Although there might have been a difficulty in synthesizing amino acid molecules in the environment on primitive Earth, the recent collection of micrometeorites in Antarctica suggests that extraterrestrial amino acids might have been delivered to primitive Earth [compare F. Brinton, C. Engrand, D. P. Glavin, J. L. Bada, M. Maurette, Origins Life Evol. Biosphere 28, 413 (1998) [ISI][Medline] ].
- The stirring condition was confirmed by measuring the through-time of the marker, vitamin B12, in the whole closed circuit under the pressurized condition, but without heating.
- After the reactor was operated for 1 hour, the pH of the circulating fluid increased from an initial pH of 2.5 to a pH of 2.9, suggesting an involvement of catabolic reactions.
- M. Nagayama, O. Takaoka, K. Inomata, Y. Yamagata, Origins Life Evol. Biosphere 20, 249 (1990) [ISI][Medline] .
- We thank A. Maeshima, Y. Ogata, and M. Sato for their assistance in performing the experiments reported here.